Keeping the Script Clean
DNA replication is a story told billions of times over in every living being, and yet almost always without error. The narrator of this story is DNA polymerase ε, an enzyme with two essential talents: one hand writes, stringing nucleotides together into a faithful copy of the genome, while the other edits, catching and excising rare mistakes before they can take root. It is this delicate balance of writing and proofreading that keeps the script of life coherent across generations of cells.
A Cancer-Linked Typo
But sometimes the editor falters. Sequencing of colorectal and, less often, endometrial cancers revealed a curious pattern: a subset of tumors carried mutations in Pol ε’s proofreading domain. Among them, one variant—POLE-P286R—stood out for the sheer weight of its consequences. Tumors with this mutation were not just error-prone; they were drenched in mutations, their genomes scarred by single-nucleotide changes at astonishing density.
At first, the explanation seemed obvious: without proofreading, mistakes simply accumulate. But when my team and I recreated the mutation in yeast, the result was far more extreme. The mutant polymerase generated errors at a pace that dwarfed even proofreading-deficient controls. This wasn’t ordinary sloppiness. It was ultra-mutagenesis, as though the enzyme had not only dropped its red pen but had begun scribbling furiously, unable to stop.
When Proofreading Fails Loudly
Looking closely at the patterns of these mutations, we saw something hauntingly familiar. The scars matched a fingerprint already known in human cancer genomes: mutational Signature 14. For years, this signature had been observed but unexplained. Now its origins were clear—it was the mark of a proofreader gone rogue.
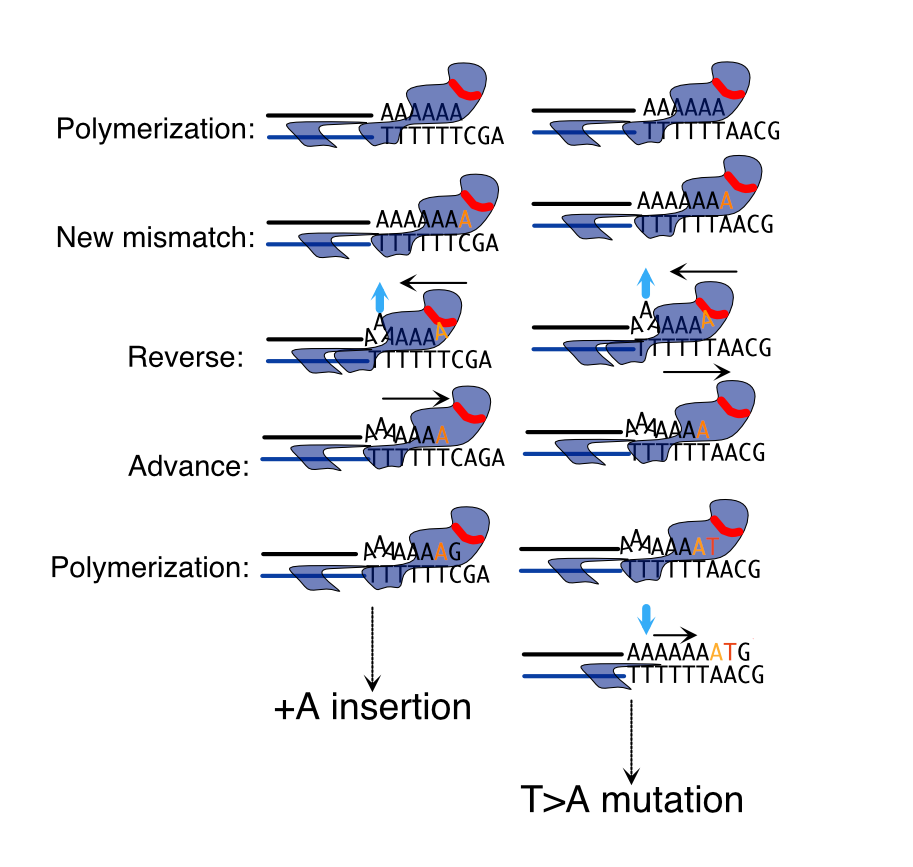
The mechanism of this collapse lies in the choreography between the polymerase’s two hands. Normally, when a wrong base is slipped into the growing DNA chain, the strand is passed into the proofreading domain, the mistake removed, and synthesis continues smoothly. In the mutants, that transfer fails. The mismatched primer strand cannot be accommodated, and the enzyme stalls. Instead of correcting the slip, it creates new wounds: characteristic T→A substitutions and small insertions, especially in stretches of repeated bases. Each one a misplaced stroke, an error left to linger.
Even more striking was the comparison with mutations in the polymerase domain itself. Though the defects lay in different hands of the enzyme, the patterns of mutagenesis were nearly indistinguishable. Whenever Pol ε malfunctions—whether in writing or editing—the genome is subjected to the same runaway instability. Yet proofreading-domain variants add their own quirks, layering unique signatures on top of the chaos.
That this story plays out identically in yeast and in humans is a reminder of how deeply conserved the mechanics of DNA replication are. The lessons drawn from a single-celled organism extend directly to the mutational landscapes of human tumors.
In the end, a single amino acid change—just one misplaced note—can send a whole system off key. Pol ε, once a careful narrator of the genome, becomes an unreliable storyteller, scattering misprints across the DNA text. For cancer cells, these mistakes provide raw material for evolution and survival. For researchers, they are the echoes of a polymerase that went off script, a warning of how fragile the balance of fidelity can be.
Read the article here