Yeast has long been a workhorse of genetics and molecular biology. Yet, despite decades of study, certain regions of its genome have remained elusive—particularly the repetitive sequences that are notoriously difficult to map and analyze. We have created a revised yeast reference genome aimed at improving read mapping in repetitive areas.”
The Challenge of Repetitive Sequences
Traditional reference genomes often collapse repetitive regions into single consensus sequences. While this simplifies assembly, it creates a major problem for read mapping: sequences derived from repeats may map ambiguously or fail to map entirely. This has hindered studies of gene families, transposable elements, rDNA arrays, and other repetitive elements that play crucial roles in genome function and evolution.
A Novel Approach: Separate Contigs for Repeats
The new yeast reference genome (SacCerRep) takes a fundamentally different approach. Instead of collapsing repeats, these bave been extractd and represented as separate contigs, each present only once. By doing this:
- Reads originating from repetitive regions now have a unique location to map to.
- Ambiguity in mapping is drastically reduced.
- Coverage and alignment metrics improve, enabling more accurate quantification of repetitive sequences.
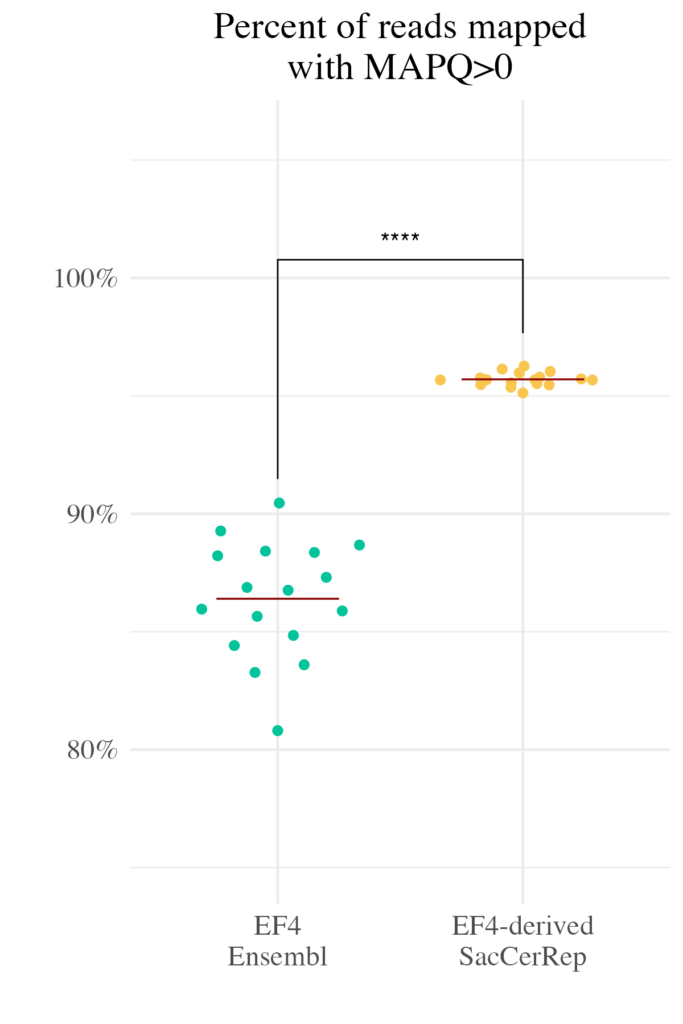
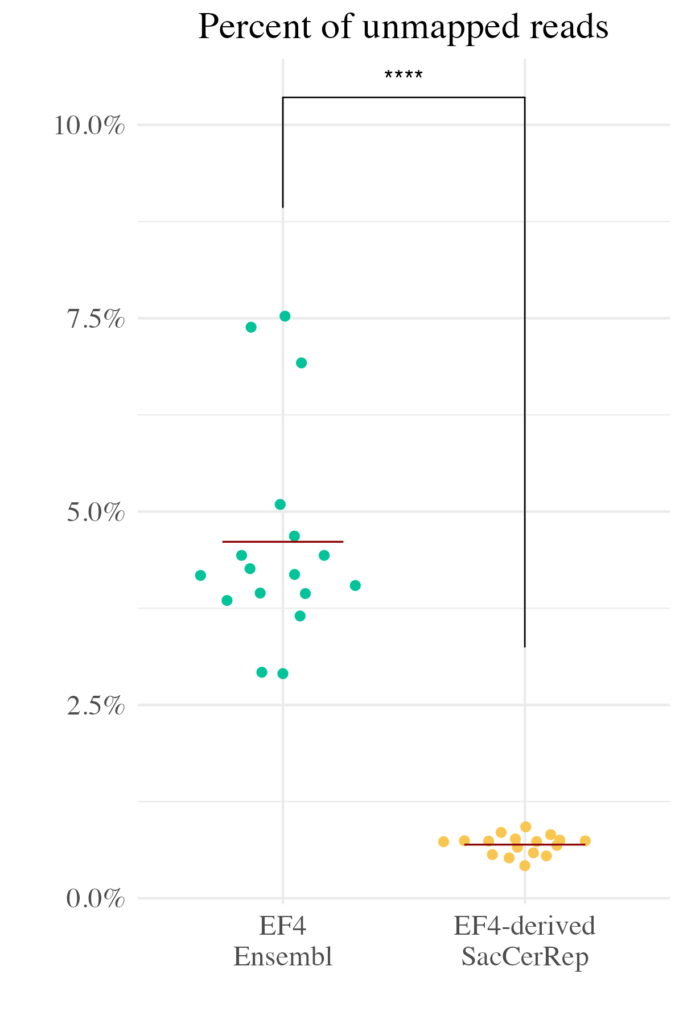
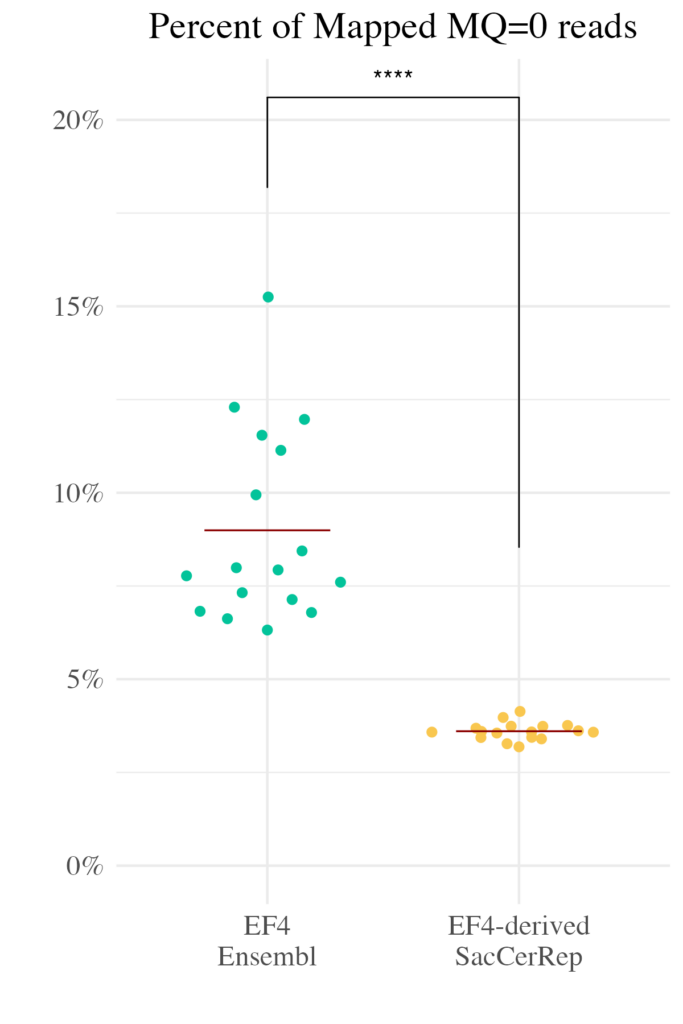
This strategy effectively “unpacks” the repetitive genome, providing researchers with a clearer, more accessible view of these previously challenging regions.
Benefits for Research
- Improved Mapping Rates: By assigning unique contigs to repetitive elements, the genome dramatically increases the proportion of reads that can be confidently mapped.
- Enhanced Resolution of Repeats: We can now study copy number variation, structural variants, and transcriptional activity in repetitive regions with unprecedented precision.
- A More Complete Genome Picture: The new reference integrates both unique and repetitive sequences without compromising assembly quality, offering a holistic view of the yeast genome.